J.ophthalmol.(Ukraine).2022;6:30-34.
|
http://doi.org/10.31288/oftalmolzh202263034 Received:14.04.2022; Accepted: 02.12.2022; Published on-line: 21.12.2022
Rare neurogenic retinal tumors in adults: morphological features and diagnostic challenges
M. V. Lytvynenko 1, V. V. Alekseeva 2, 3, V. V. Gargin 2, 3, N. V. Neskoromna 1, O. L. Koshelnyk 1, O. V. Artemov 4 1 Odesa National Medical University; Odesa (Ukraine) 2 Kharkiv National Medical University; Kharkiv (Ukraine) 3 Kharkiv International Medical University; Kharkiv (Ukraine) 4 SI "The Filatov Institute of Eye Diseases and Tissue Therapy of the NAMS of Ukraine"; Odesa (Ukraine) TO CITE THIS ARTICLE: Lytvynenko MV, Alekseeva VV, Gargin VV, Neskoromna NV, Koshelnyk OL, Artemov OV. Rare neurogenic retinal tumors in adults: morphological features and diagnostic challenges. J.ophthalmol.(Ukraine).2022;6:30-34. http://doi.org/10.31288/oftalmolzh202263034
Background: The histological diagnosis of neurogenic tumors remains a challenge, which may be indicated particularly by the fact that new entities appeared in the new edition of the World Health organization (WHO) classification. Purpose: To review the histomorphologic and immunohistochemic features of rare variants of neurogenic ocular (retinal) tumors in adults. Material and Methods: Six rare ocular tumors were selected for the study from all clinical material submitted for pathohistological examination from 2017 to 2020 based on the presence of morphological evidence of neurogenic differentiation. Results: The study sample of six rare neurogenic retinal tumors in adults was conventionally divided into three types: (1) retinal tumors immunohistochemically similar to cellular ependymoma, but histologically similar to retinoblastoma; (2) tumors showing no histological pattern characteristic for dictyoma, but the immunohistochemical features of neuroepithelial differentiation; and (3) tumors showing histological patterns similar to medulloepithelioma, but the immunohistochemical features of glial markers. Conclusion: Obviously, when dividing these tumors into histogenetic groups, not only the histological structure and immunohistochemical profile, but also tumor location and typical patient age should be taken into account.
Keywords: ocular tumors, histopathology, immunohistochemistry
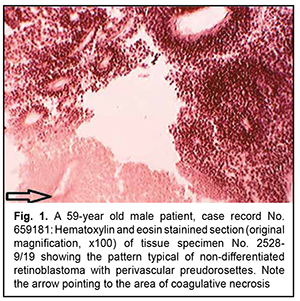
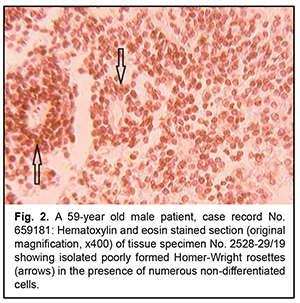
Introduction As the histological diagnosis of neurogenic tumors remains a challenge, new entities appeared in the new edition of the World Health organization (WHO). Histological variants were added if there was evidence of a different age distribution, location, genetic profile or clinical behavior. In the course of the revision process of the classification, the being of entities can be clarified; information that is spread over (at least) two entities can be fused together; and/or some entities (or variants) that no longer have relevance can be deleted from the classification [1]. These classification problems are also pertinent to neurogenic ocular tumors. Of these tumors, the most commonly encountered are retinoblastoma and dictyoma, which occur in early childhood (mostly, before 5-7 years of age). The most common location of dictyoma is the anterior segment of the eye (anteriorly to the ora serrata). Consequently, diagnosing tumors showing (a) the histological patterns characteristic for the above neoplasms, in elderly and/or (b) atypical location, is a challenge. In addition, age of the patient and tumor location should be taken in account when differentiating between neurogenic tumors [2]. The purpose of the study was to review the histomorphologic and immunohistochemic features of rare variants of neurogenic ocular (retinal) tumors in adults. Material and Methods Six rare ocular tumors with signs of neurogenic differentiation were selected for the study from all clinical material submitted for pathohistological examination from 2017 to 2020. These tumors were selected on the basis of (a) the presence of histological patterns typical of neurogenic (retinal) tumors and (b) the age that contrasted significantly with the typical age distribution encountered in these tumors. The material was processed in a routine histological manner. Histological sections were stained with hematoxylin and eosin, and paraffin blocks were sent for immunohistochemical analysis. Immunohistochemical markers were selected on the basis of the algorithm for the study of neurogenic brain tumors. Results Rare variants of neurogenic ocular (retinal) tumors were divided into types based on the types of below mentioned histological patterns. Type I, neurogenic retinoblastoma, was found in three patients (aged between 59 to 62 years) of the study sample. Although all six neoplasms were identified clinically as uveal melanomas, macroscopic examination showed no pigment or a nodule typical of these tumors, but loose tumor tissue and small flake-like tumor aggregates not associated with the primary tumor, a feature more typical of retinoblastoma. Micrographs in figures 1 and 2 show typical histomorphological patterns of retinoblastoma (lymphocyte-like round cells that form a perivascular preudorosette; areas of coagulative necrosis with a hardly noticeable tendency to accumulation of calcium salts; and isolated true rosettes).
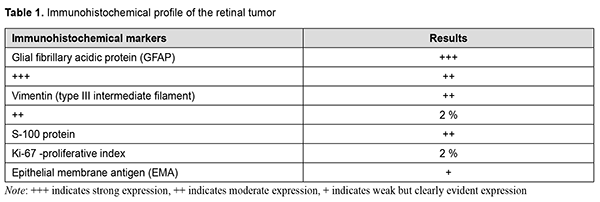
Although histomorphological examination revealed some characteristics consistent with retinoblastoma, patient’s age was not characteristic, and tumor pathology showed no Flexner-Wintersteiner rosettes that would be characteristic of retinoblastoma, leading to diagnostic uncertainty. Indeed, there are scarce reports of retinoblastoma in adults [3-5]. It is noteworthy that nodular tumor growth was common in all our observations, and involved predominantly the anterior and posterior equatorial regions. In addition, necroses were associated with epichoroidal growth; they, however, were rare in cases in which choroidal growth was more common than epichoroidal growth, which is also typical of retinoblastoma. The presence of necroses not only indicates the similarity between these tumors and retinoblastoma with regard to histomorphological picture, but also indirectly indicates the similarity with regard to histogenetic sources. There are grounds to believe that the tapetal retinal region is a source of growth both for these tumors and retinoblastoma. The tapetal retinal pigment epithelium (RPE) and the tapetal photoreceptors have no specific stromal and vascular area. Consequently, the tumor parenchyma will be susceptible to necrosis until the tumor growth is not supported by neovascularization from the choroid. One of these retinoblastoma-like tumors was immunohistochemically positive for the markers listed in table 1, and negative for Cytokeratin MNF-116, Neurofilaments, Sinaptophysin, CD-45, CD-56, CD-99, and HMB-45. Similar to the algorithm for immunohistochemical analysis of CNS tumors, the tumor was initially classified as a grade 2 cellular ependymoma, because, similar to a grade 2 cellular ependymoma, the tumor was positive for glial marker GFAP and negative for neuronal differentiation markers such as neuron-specific enolase, neurofilament, and synaptophysin.
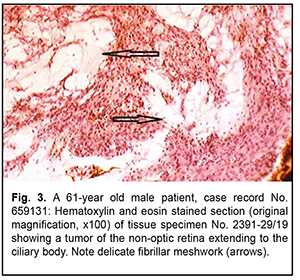
Naturally, the immunohistochemical overlap between the tumor and a cellular ependymoma shall not be sufficient for transferring the diagnosis to ocular tumors, all the more because cellular ependymoma is an entity that has been revised in the last edition of the WHO classification due to its ambiguity. In addition, based on the location and histological features of the tumor, it seems most reasonable to compare it not with tumors of the CNS, but with retinoblastoma. It has been reported that, immunogistochemically, retinoblastoma is positive for neuronal differentiation markers such as neuron-specific enolase, neurofilament, and synaptophysin, whereas the questionable tumor was negative for these markers. However, on rare occasions, retinoblastoma may exhibit signs of glioblast differentiation, demonstrating positivity to the same glioblast differentiation markers as Muller cells [6]. Therefore, taking into account an atypical patient age range, a partial overlap in immunohistochemical profiles and a histomorphological similarity between the tumor in question and retinoblastoma, the former tumor may be recognized as a special variant of retinoblastoma-like retinal tumor. A comparative molecular genetic analysis of the tumor in question and retinoblastoma will allow determining the degree of identity between them. Of the tumors sporadically verified as medulloepithelioma in a highly atypical patient range, we wish to draw attention to the variants exhibiting special histological patterns and some immunohistochemical nuances. The two examined ocular tumors of the non-optic retina extended mostly to the ciliary body, showing no pattern typical of dictyoma, but the patterns typical of neuroepithelial tumors. Imminohistochemically, they were positive for neuron-specific enolase and had low expression of GFAP. Nevertheless, histologically, they showed a tendency to the formation of the retinal neuroglial substance typical of glial tumors (note arrows in Fig. 3).
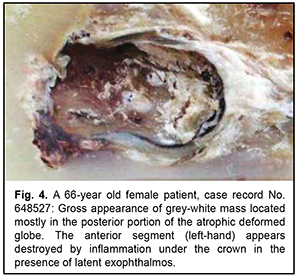
Another primary neurogenic retinal tumor not associated with the ciliary body epithelium showed the formation of a delicate fibrillar meshwork having an appearance similar to the cerebral neuropil. There were sporadic reports of retinal tumors (“astrocytoma”) histologically similar to the above tumor, mostly in the pre-immunohistochemical era. In this connection, we present a unique observation of the retinal tumor in a 64-year-old woman. It was difficult to determine disease duration, because the process occurred in the atrophic eye which was covered by crown prosthesis. The patient reported that the eye had been blind since childhood. Only after a marked exophthalmos developed, apparent inflammatory changes were found under the removed crown, and computed tomography revealed a tumor nodule as large as 3 cm in the orbit, the nodule being fused with bony structures. The patient had a partial exenteration due to a clinical picture of malignant orbital tumor. Fig. 4 shows loose grey-white tissue at the time of exenteration as a large mass filling a significant portion of the atrophic deformed globe that measured as much as 1.2 cm in the largest dimension.
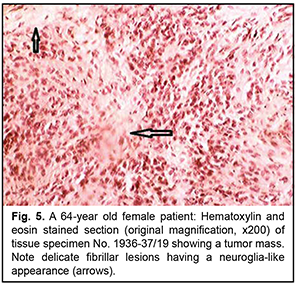
The orbit contained a grey-yellow nodule measuring 2.5 x 2.0 cm and located separately from the globe; the nodule was tightly adhered to the periosteum and adjacent orbital soft tissues. Histologically, the tumor process in the ocular cavity was identical to that in the orbital nodule (Fig. 5).
Similar to the algorithm for immunohistochemical analysis of CNS tumors, the tumor was classified as a grade 2 glioblastoma. However, histologically, the process is more similar to the tumors described in the literature as ocular astrocytoma. This tumor is histologically similar to that qualified as retinal astrocytoma by Shields and colleagues [7]. Interestingly, that Vit [8] quotes the histological findings of the cases reported by Shields and colleagues [7] as a rare example of ocular tumor. Ocular astrocytomas, by analogy to astrocytomas in the brain, are not uncommonly believed to be benign. Obviously, the clinical consequences can be favorable, since the invasion to the outer ocular coats is commonly absent due to early diagnosis and enucleation. However, the case reported in the current paper raises a question whether the above conclusion is correct. In this case, after the tumor process became uncontrolled due to the presence of the crown, there developed an extrabulbar tumor nodule associated with the orbital wall. This required exenteration, a disabling surgery, which is rarely performed even in cases of ocular melanoma. All this things prevent us from classifying this tumor as benign. Here the diagnosis of glioblastoma is based on immunohistological markers (i.e., an immature tumor of glial cells) more adequately represents the nature of this tumor than the diagnosis of astrocytoma. However, we must agree that the histological features do not show the biological potential of this tumor. Indeed, the tumor is composed of relatively monomorphic round and spindle-shaped cells (without mitotic figures) which are alternated with the areas of glial stroma (these areas are shown by arrows in Fig. 4). Discussion The cases of neuroglial tumors reported here need to be reviewed in light of the 2016 CNS WHO and the fifth edition of the WHO Classification of Tumors of the CNS, published in 2021 [9]. Auxiliary imaging techniques, both conventional and novel, may substantially improve diagnostic assessment [10-13]. These data are especially valuable given complex transformations of melanocytes, epithelium and neural tissue of the orbital region [14-16]. The rare neuroglial tumors in adults reported here may be conventionally divided into three types: (1) retinal tumors histologically similar to retinoblastoma, but having some immunohistochemical features; (2) tumors of the non-optic retina showing no immunohistochemical pattern characteristic for medulloepithelioma or dictyoma, but the patterns typical of neuroepithelial tumors; and (3) retinal tumors showing histological patterns similar to posterior-segment ocular tumors of astroglial origin positive for glial markers. The study demonstrates that neurogenic ocular (retinal) tumors in adults vary in histological and immunohistochemical patterns, and it is difficult to develop their diagnostic criteria because these tumors are rather rare. A rare ocular neuroglial tumor should be diagnosed taking into account its immunohistochemical profile. In fact, this requirement renders a diagnosis of a rare ocular neuroglial tumor established without taking into account the immunohistochemical profile null and void. Obviously, these descriptions are useful in the context of their retrospective evaluation, but it is too early to make final conclusions on developing a new classification for rare tumors. Now we are just at the stage of initial systematization and thus have to apply brain tumor algorithms in immunohistochemical studies. However, even our sparse observations reported in this paper demonstrate that, with regard to this matter, analogies between brain tumors and ocular neuroglial tumors are of somewhat limited value. Conclusion Our histomorphological and immunohistochemical analysis of rare intraocular tumors of retinal origin allowed dividing them conventionally into three close histogenetic groups, with the diagnostic assessment being based on comparing these tumors with neurogenic brain tumors with regard to histological and immunohistochemical patterns. Obviously, when dividing these tumors into histogenetic groups, not only the histological structure and immunohistochemical profile, but also tumor location and typical patient age should be taken into account.
References 1.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Bernd AJ, et al. The 2007 WHO Classification of tumours of the central nervous system. Acta Neuropathol. 2007; 114(2):97–109. 2.Gupta A, Dwivedi T. A simplified overview of World Health Organization classification update of central nervous system tumors 2016. J Neurosci Rural Pract. Oct-Dec. 2017; 8(4):629-641. 3.Biswasm J, Manim B, Shaunmugen M. Retinoblastoma in adults: report of three cases and review of the literature. Surv Ophthalmol. 2000 Mar-Apr;44(5):409-14. 4.Mackley R.A. Retinoblastoma in a 52-year old man. Arch Ophthalmol. 1963 Mar;69:325-7. 5.Takahahi T, Namura S, Inoue M., Isayama Y, Sashikata T. Retinoblastoma in a 26-year old adult. Ophthalmology. 1983 Feb;90(2):179-83. 6.Vit VV. [Pathology of the eye, ocular adnexa and orbit: a two-volume monograph]. Vol. 2. Odesa: Astroprint; 2019. Russian. 7.Shields JA, Eagle RC, Jr, Shields CL, Marr BP. Aggressive retinal astrocytomas in four patients with tuberous sclerosis complex. Trans Am Ophthalmol Soc. 2004;102:139-47; discussion 147-8. 8.Vit VV. [Visual system tumors: a two-volume monograph]. Vol. 2. Odesa: Astroprint; 2009. Russian. 9.Louis DN, Perry A, Wesseling P, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021;23(8):1231-1251. 10.Schabadasch A. Intramurale nervengeflechte des darmrohrs. Z Zellforsch Mikrosk Anat. 1930;10:320–85. 11.Gargin V, Radutny R, Titova G, Bibik D, Kirichenko A, Bazhenov O. Application of the computer vision system for evaluation of pathomorphological images. In: Proceedings of the IEEE 40th International Conference on Electronics and Nanotechnology, ELNANO 2020. 22-24 April 2020, Kyiv, Ukraine. 469-73. 12.Garancher A, Lin CY, Morabito M, et al. NRL and CRX Define Photoreceptor Identity and Reveal Subgroup-Specific Dependencies in Medulloblastoma. Cancer Cell. 2018;33(3):435-449.e6. 13.Stenzinger A, Alber M, Allgäuer M, et al. Artificial intelligence and pathology: From principles to practice and future applications in histomorphology and molecular profiling. Semin Cancer Biol. 2022 Sep;84:129-43. 14.Sulym H, Lyndin M, Sulym L, et al. Detection of melanin in the rat skin. Pol Merkur Lekarski. 2022;50(295):21-4. 15.Lytvynenko M, Shkolnikov V, Bocharova T, Sychova L, Gargin V. Peculiarities of proliferative activity of cervical squamous cancer in HIV infection. Georgian Med News. 2017;(270):10-15. 16.Goto H, Yamakawa N, Komatsu H. Histopathology and immunohistochemistry of choroidal melanocytoma demonstrated by local resection: A case report. Am J Ophthalmol Case Rep. 2021;23:101147.
Disclosures Received 14.04.2022 Accepted 02.12.2022 Corresponding author: Артьомов Олександр Валентинович – art_onkol@ukr.net Disclaimer. The authors declare that the opinions expressed in the article are their own and not the official positions of the institution. Conflict of interest. The authors declare that they have no conflicts of interest that could influence their opinion on the subject matter or materials described and discussed in this manuscript. Sources of support: none.
|