J.ophthalmol.(Ukraine).2021;2:40-45.
|
http://doi.org/10.31288/oftalmolzh202124045 Received: 10 July 2020; Published on-line: 19 April 2021 Results and possible prospects of genetic technology in ophthalmology (literature review). Part 2. N. A. Gavrilova, O. Ie. Tishchenko, A. V. Zinov’eva A.I. Evdokimov Moscow State University of Medicine and Dentistry; Moscow (Russian Federation) E-mail: aleksandra.r@live.ru TO CITE THIS ARTICLE: Gavrilova NA, Tishchenko OIe, Zinov’eva AV. Results and possible prospects of genetic technology in ophthalmology (literature review). Part 2. J.ophthalmol.(Ukraine).2021;2:40-45. http://doi.org/10.31288/oftalmolzh202124045 The emergence of fundamentally novel technological solutions in the field of gene therapy today, the formation of the priority and the development of genetic technologies create serious prerequisites for the beginning of a new Fusion era in ophthalmology in the near future. This review, in its second part, presents the results of fundamental and clinical studies on the use of genetic therapeutic strategies – gene replacement, gene suppression, genomic editing using CRISPR / Cas9 technology which have been used in ophthalmology over the past several years. Keywords: small interfering RNAs, antisense nucleotides, CRISPR, gene therapy, retina
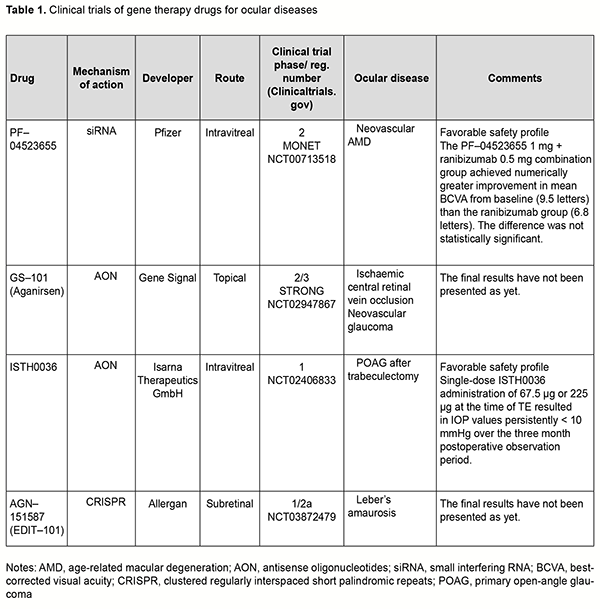
Gene replacement strategy Ali and colleagues (2000) [1] used the approach of replacement of the mutated Prph2 gene for the treatment of autosomal dominant retinitis pigmentosa in mice. The gene PRPH2 encodes for the peripherin-2 protein, is involved in cell interaction with the extracellular matrix and other cells, and stabilizes membrane discs of photoreceptor outer segments. Mutations in Prph2 cause failures of photoreceptor formation and arrangement, and have been shown to result in a variety of retinal degenerative diseases like macular degeneration, Stargardt disease, Leber’s amaurosis, and punctata albescens retinopathy. Subretinal injection of recombinant adeno-associated virus (AAV) encoding a Prph2 transgene resulted in restoration of photoreceptor structure and function, but the duration of functional improvement was not long. Mutation of a receptor tyrosine kinase gene, Mertk, in the Royal College of Surgeons (RCS) rat results in defective phagocytosis of photoreceptor outer segments by the retinal pigment epithelium (RPE). MER tyrosinase kinase protooncogene (MERTK) is a member of the Tyro3, Axl, and MerTK (TAM) family of receptors. Subretinal injection of a recombinant replication-deficient adenovirus encoding rat Mertk in the RCS rat resulted in restoration of RPE phagocytosis at sites adjacent to the injection site as well as a delay in photoreceptor degeneration [2]. Other studies reported on AAV-mediated [3] and LV-mediated [4] MERTK gene transfer, with restoration of both photoreceptors and phagocytic capacity of the RPE and increased electroretinographical (ERG) activity for 9 weeks after AAV-mediated treatment and 4 months after LV-mediated treatment for the RCS rat model of retinitis pigmentosa. There have been several reports on gene replacement approach to the treatment of congenital Leber’s amaurosis. Subretinal injections of AAV.RPE65 were performed for RPE65 gene replacement in Rpe65 knockout (Rpe65−/−) mice, resulting in functional restoration and delay in degeneration of photoreceptors. Optimized AAV2-mediated gene transfer through subretinal injection of AAV2.RPE65 resulted in increased ERG activity and improved visual acuity in Rpe65 knockout mice for at least 3 months, with no report on degeneration of photoreceptors [5]. The findings of the study led to the development of Luxturna (Spark Therapeutics, Philadelphia, PA), a pharmaceutical for treating congenital Leber’s amaurosis (see the first part of the review for more details). Mutations in the ABCA4 gene cause Stargardt's disease (STGD), a form of autosomal recessive juvenile macular degeneration. Subretinal delivery of rAAV2/5-CMV-Abca4in a murine model of STGD resulted in significant correction of lipofuscin levels, RPE abnormalities, and retinal function for a period of up to 5 months [6]. Gene silencing strategy Gene silencing can occur at the transcription level or post-transcription level. Gene silencing at the transcription level is a result of histone modification in heterochromatin, leading to unavailability of particular DNA loci for transcriptional apparatus (RNA-polymerase) and transcription factors. Gene silencing at the post-transcription level is a result of degradation of mRNA of particular genes. Degradation of mRNA inhibits the translation to the complementary protein. RNA interference (RNAi) is a widely used post transcriptional silencing mechanism for suppressing expression of the target gene. Various antisense inhibitors (ribozymes, miRNA inhibitors, and antisense oligonucleotides) are employed for this purpose. Ribozymes Ribozymes (Rz) are small RNA structures with high catalytic activity and specificity. In recent years, there have been advances in the development of ribozymes with improved characteristics and their applications for inhibiting gene expression. The ribozymes designed to cleave the P23H and S334Ter mutant RNA selectively were employed for treating autosomal–dominant retinitis pigmentosa (adRP) [7, 8]. In addition, mutation-independent hammerhead ribozymes targeting rhodopsin and peripherin have been screened in vitro, and a number of extremely efficient ribozymes identified subsequent to detailed kinetic analyses, suggesting that these ribozymes may provide mutation-independent methods of treating adRP [9, 10]. Gorbatyuk and colleagues [11] reported that subretinally injected AAV2-Rz397 led to significant (greater than or equal to 50%) reduction of rhodopsin mRNA and protein in rhodopsin knockout hemizygous (RHO+/-) mice [11]. In another study by Gorbatyuk and colleagues [12], a ribozyme targeting dog, mouse, human but not rat RHO mRNA was designed and tested in vitro. Rz525 driven by the mouse opsin proximal promoter was inserted in plasmids with AAV 2 terminal repeats and packaged in AAV serotype 5 capsids. AAV-Rz525 was injected subretinally into the right eyes of P23H rat pups. Left eyes were injected with virus expressing GFP from the identical promoter. RT-PCR analysis revealed 46% reduction of transgenic (mouse) RHO mRNA in right eyes relative to left eyes and no change in rat RHO mRNA. Before they can be used for human gene therapy, ribozymes such as Rz525 may need to be coupled with a ribozyme resistant RHO gene as a replacement for endogenous rhodopsin. It is probably prudent to deliver the ribozyme and the replacement gene in one package, i.e., using the same AAV vector. Short interfering RNAs (siRNAs) Short interfering RNAs (siRNAs) are 20–25 nucleotide double-stranded RNA (dsRNA) molecules that target genes at expression level and inhibit the synthesis of pathological proteins [13, 14, 15]. An RNA-mediated mechanism for inhibiting the expression of specific genes — the RNA interference (RNAi) pathway — is especially promising for retinovascular disorders, and relevant clinical trials are underway. In a phase II clinical trial (MONET) [16] evaluating treatment for neovascular AMD, the combination of intravitreal siRNA PF-04523655 (Quark; licensed to Pfizer) with ranibizumab led to an average gain in BCVA that was more than with ranibizumab monotherapy, and no safety concerns were identified. Antisense oligonucleotide therapy Of the various gene therapies currently available, antisense oligonucleotide (AON) therapy is increasingly being developed in ophthalmology and of special interest. The rationale of the antisense strategy is to inhibit synthesis of deleterious proteins by blocking the function of their corresponding mRNA. Antisense oligonucleotides are short strands of synthetic nucleic acid which bind target RNA by complementary base pairing. In recent years, numerous preclinical studies on AON therapy for inherited retinal disorders, retinoblastoma, corneal dystrophy and neovascularization [17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27] have being conducted. GS-101 is eye drops of aganirsen, a novel AON preventing insulin receptor substrate-1 (IRS–1) mRNA expression, with a completed phase III clinical trial. The drug has been shown to inhibit corneal neovascularization after keratoplasty and stromal keratitis and to prevent neovascular glaucoma in ischemic central vein occlusion (a phase II/III clinical trial for the Study of a Topical Treatment of Ischaemic Central Retinal Vein Occlusion to Prevent Neovascular Glaucoma - the STRONG study) [28, 29]. A first-in-human phase I study of ISTH0036, an antisense oligonucleotide selectively targeting transforming growth factor beta 2 (TGF-β2) has been conducted in subjects with open-angle glaucoma undergoing glaucoma filtration surgery [30]. CRISPR/Cas9–mediated genome editing The clustered regularly interspaced short palindromic repeats/CRISPR-associated proteins (CRISPR/Cas9) technology evolves from a bacterial immune system and represents a new generation of targeted genome editing technology. The basis for this technology is segments of bacterial DNA called CRISPRs interspaced by fragments of foreign DNA (spacers) with remnant viral traces. The CRISPR arrays allow the bacteria to "recollect" the viruses. If a virus attacks again, the bacteria produce RNA segments from the CRISPR arrays to specifically target the virus' DNA. Studies on genome editing in cells using the CRISPR/Cas9 system began in 2013. Jennifer Doudna and Emmanuel Charpentier demonstrated that CRISPR RNA could be сonstructed, with a particular DNA fragment replaced by the sequence specified by the researcher. The Cas9 protein recognizes and binds to the trans-activating crRNA, and the crRNA functions as a guide so that Cas9 can recognize and cleave the DNA target sequence. This system enables cutting out a ‘pathologic’ genome locus and replacing it by a ‘healthy’ genome locus. The cell will not die from DNA break, as it will be corrected by a healthy copy of the mating chromosome due to the natural process of DNA repair. A number of studies using the CRISPR/Cas9 genome editing tool have been performed in ophthalmology in recent years. CRISPR/Cas9 mediated knockout animal models of retinitis pigmentosa and retinoblastoma [31, 32] have been presented, and gene therapy strategies for the treatment of inherited diseases such as corneal dystrophy, congenital Leber’s amaurosis, X-linked retinoschisis, and wet age-related macular degeneration, developed [33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44]. Of note are studies employing CRISPR/Cas9-mediated genome editing for CRISPR-based suppression of VEGF-A, blocking VEGF-R2 and angiogenesis in animal models of oxygen-induced retinopathy and laser-induced choroidal neovascularization [45, 46, 47]. Findings of a study on CRISPR/Cas9-mediated gene replacement in the fungal keratitis pathogen, Fusarium solani var. petroliphilum give promise that genome editing can be used for the treatment of persistent ocular surface infection [48]. Safe delivery of CRISPR/Cas endonucleases remains one of the major barriers to the widespread application of in vivo genome editing. With adeno-associated virus (AAV)-mediated CRISPR/Cas genome editing in the retina, the CRISPR/Cas construct will be maintained in the retina for an extended period, impede cell function and induce immune responses. Nanodiamonds (NDs) are considered to be relatively safe carbon nanomaterials used for the transmission of DNA, proteins and drugs [49]. Yang and colleagues [49] used nanodiamonds as the carriers of CRISPR-Cas9 components designed to introduce the mutation in RS1 gene associated with X-linked retinoschisis (XLRS). Rs1 gene editing in mouse retinas resulted in several pathological features typical for XLRS. Li and colleagues [50] designed a self-destructing "kamikaze" CRISPR/Cas system that disrupts the Cas enzyme itself following expression. Their data suggested that a self-destructive "kamikaze" CRISPR/Cas system could be used as a robust tool for genome editing in the retina, without compromising on-target efficiency. Another barrier to the widespread application of CRISPR-mediated genome editing is the large size of Cas9 endonuclease. The commonly used Streptococcus pyogenes Cas9 is large and may impose packaging problems for delivery by a single AAV, but delivery using a combination of two AAV vectors has been reported to be less efficient than delivery by a single AAV vector. One solution to the carrying capacity issue is to use smaller Cas9 orthologs, and orthologs from the CRISPR systems in Campylobacter jejuni (CjCas9) and Staphylococcus aureus (SaCas9) are being considered for this purpose. Unexpected off-target mutations after CRISPR–Cas9 editing are the most serious limitation for applications of CRISPR–Cas9 systems. Schaefer and colleagues [51] used CRISPR-Cas9 for sight restoration in blind rd1 mice by correcting a mutation in the Pde6b gene. They found that CRISPR gene correction introduces an unexpectedly high number of mutations in a mouse model of gene therapy. In 2019, the BRILLIANCE trial became the first clinical trial to deploy the CRISPR–Cas9 technique directly in the body. In BRILLIANCE, gene editing is used to delete a mutation in the gene CEP290 that is responsible for Leber’s congenital amaurosis 10 (LCA10). The conventional СRISPR–Cas9 genome-editing system has turned out to be not perfect for every situation, and one limitation of the system is the potential for off-target modifications, i.e., the modification of sequences similar to the intended target sequence. Therefore, the genome-editing technology continues to evolve, with the main aim of excluding off-target effects. Novel genome-editing technologies (prime editing, transposon genome editing and DNA shredder technique) Prime editing A prime editing complex consists of a prime editor protein fused to a reverse transcriptase domain and complexed with a prime editing guide RNA (pegRNA) [52]. Unlike CRISPR edits, the prime editing Cas9, a nickase, cuts a single DNA strand. The precise character of the editing method reduces the off-target effects seen with CRISPR-Cas9. DNA transposons, or "jumping genes," always move on their own, inserting or excising themselves from the genome by means of a so-called "cut and paste" mechanism. An RNA molecule is formed during transcription; the transposase is built in the ribosome using RNA instructions, returns to the nucleus, recognizes a particular DNA sequence (the transposone), cleaves the ends of the transposon and also cleaves target sites where the element is to be inserted. Once the transposon is bound into its new position, gaps that are left in the DNA sequence are filled through the synthesis of nucleotides. Hernandez and colleagues [53] evaluated the effect of ex-vivo cell-based gene therapy using the Sleeping Beauty transposon system in laser-induced choroidal neovascularization (CNV), with transfected primary cells injected into the subretinal space. There was a significant increase in human pigment epithelium-derived factor (PEDF) and the PEDF/VEGF ratio with transfected RPE cells and a reduction in CNV area.
Conclusion Therefore, advanced gene therapy strategies like gene suppression using RNA interference, antisense oligonucleotides, CRISPR/Cas9-mediated genome editing, and, especially, novel technologies like prime editing and transposon editing offer wide opportunities for applications in ophthalmology.
References 1.Ali RR, Sarra GM, Stephens C, Alwis MD, Bainbridge JW, Munro PM, et al. Restoration of photoreceptor ultrastructure and function in retinal degeneration slow mice by gene therapy. Nat Genet. 2000;25(3):306‐10. 2.Vollrath D, Feng W, Duncan JL, Yasumura D, D'Cruz PM, Chappelow A, et al. Correction of the retinal dystrophy phenotype of the RCS rat by viral gene transfer of Mertk. Proc Natl Acad Sci USA. 2001;98(22):12584‐9. 3.Smith AJ, Schlichtenbrede FC, Tschernutter M, Bainbridge JW, Thrasher AJ, Ali RR. AAV–Mediated gene transfer slows photoreceptor loss in the RCS rat model of retinitis pigmentosa. Mol Ther. 2003;8(2):188‐95. 4.Tschernutter M, Schlichtenbrede FC, Howe S, Balaggan KS, Munro PM, Bainbridge JWB, et al. Long‐term preservation of retinal function in the RCS rat model of retinitis pigmentosa following lentivirus‐mediated gene therapy. Gene Ther. 2005;12694–701. 5.Bennicelli J, Wright JF, Komaromy A, Jacobs JB, Hauck B, Zelenaia O, et al. Reversal of blindness in animal models of leber congenital amaurosis using optimized AAV2–mediated gene transfer. Mol Ther. 2008;16(3):458‐465. 6.Allocca M, Doria M, Petrillo M, Colella P, Garcia–Hoyos M, Gibbs D, et al. Serotype–dependent packaging of large genes in adeno–associated viral vectors results in effective gene delivery in mice. J Clin Invest. 2008;118(5):1955‐1964. 7.Drenser KA, Timmers AM, Hauswirth WW, Lewin AS. Ribozyme–targeted destruction of RNA associated with autosomal–dominant retinitis pigmentosa 5. Invest Ophthalmol Vis Sci. 1998;39:681–9. 8.Lewin AS, Drenser KA, Hauswirth WW, Nishikawa S, Yasumura D, Flannery JG, LaVail MM. Ribozyme rescue of photoreceptor cells in a transgenic rat model of autosomal dominant retinitis pigmentosa 4. Nat Med. 1998;4:967–71. 9.O'Neill B, Millington–Ward S, O'Reilly M, Tuohy G, Kiang AS, Kenna PF, et al. Ribozyme–based therapeutic approaches for autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2000;41(10):2863‐2869. 10.Sullivan JM, Pietras KM, Shin BJ, Misasi JN. Hammerhead ribozymes designed to cleave all human rod opsin mRNAs which cause autosomal dominant retinitis pigmentosa 1. Mol Vis. 2002;8:102–13. 11.Gorbatyuk MS, Pang JJ, Thomas J Jr., Hauswirth WW, Lewin AS. Knockdown of wild–type mouse rhodopsin using an AAV vectored ribozyme as part of an RNA replacement approach. MolVis. 2005;11:648–56. 12.Gorbatyuk M, Justilien V, Liu J, Hauswirth WW, Lewin AS. Preservation of photoreceptor morphology and function in P23H rats using an allele independent ribozyme. Exp Eye Res. 2007;84(1):44‐52. 13.Corydon TJ. Antiangiogenic Eye Gene Therapy. Human Gene Therapy. 2015;26(8):525–37. 14.Garba AO, Mousa SA. Bevasiranib for the treatment of wet, age–related macular degeneration. Ophthalmol Eye Dis. 2010;2:75–83. 15.Guzman–Aranguez A, Loma P, Pintor J. Small–interfering RNAs (siRNAs) as a promising tool for ocular therapy. Br J Pharmacol. 2013;170(4):730–47. 16.Nguyen QD, Schachar RA, Nduaka CI, Sperling M, Klamerus KJ, Chi–Burris K, et al; MONET Clinical Study Group. Evaluation of the siRNA PF–04523655 versus ranibizumab for the treatment of neovascular age–related macular degeneration (MONET Study). Ophthalmology. 2012;119(9):1867–73. 17.Chau VQ, Hu J, Gong X, Hulleman JD, Ufret–Vincenty RL, Rigo F, et al. Delivery of Antisense Oligonucleotides to the Cornea. Nucleic Acid Therapeutics. 2020; 18.Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat Med. 2019;25(2):225–8. 19.Collin RW, Garanto A. Applications of antisense oligonucleotides for the treatment of inherited retinal diseases. Curr Opin Ophthalmol. 2017;28(3):260–6. 20.Gerard X, Garanto A, Rozet JM, Collin R.W. Antisense Oligonucleotide Therapy for Inherited Retinal Dystrophies. Adv Exp Med Biol. 2016;854:517‐24. 21.Hu J, Rong Z, Gong X, Zhou Z, Sharma VK, Xing C, et al. Oligonucleotides targeting TCF4 triplet repeat expansion inhibit RNA foci and mis–splicing in Fuchs' dystrophy. Hum Mol Genet. 2018;27(6):1015–26. 22.Hu J, Shen X, Rigo F, Prakash TP, Mootha VV, Corey DR. Duplex RNAs and ss–siRNAs Block RNA Foci Associated with Fuchs' Endothelial Corneal Dystrophy. Nucleic Acid Ther. 2019;29(2):73‐81. 23.Moore SM, Skowronska–Krawczyk D, Chao DL. Emerging Concepts for RNA Therapeutics for Inherited Retinal Disease. Adv Exp Med Biol. 2019;1185:85‐89. 24.Rocha EM, Nominato LF, Reinach PS. Re: Cursiefen et al.: Aganirsen antisense oligonucleotide eye drops inhibit keratitis–induced corneal neovascularization and reduce need for transplantation: the I–CAN study. Ophthalmology. 2015;122(5):e28. 25.Sangermano R, Garanto A, Khan M, Runhart EH, Bauwens M, Bax NM, et al. Deep–intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet Med. 2019;21(8):1751–60. 26.Yang G, Fu Y, Zhang L, Lu X, Li Q. miR106b regulates retinoblastoma Y79 cells through Runx3. Oncol Rep. 2017;38(5):3039–43. 27.Zarouchlioti C, Sanchez–Pintado B, Hafford Tear NJ, et al. Antisense Therapy for a Common Corneal Dystrophy Ameliorates TCF4 Repeat Expansion–Mediated Toxicity. Am J Hum Genet. 2018;102(4):528–39. 28.Cursiefen C, Viaud E, Bock F, Geudelin B, Ferry A, Kadlecová P, Lévy M, et al. Aganirsen antisense oligonucleotide eye drops inhibit keratitis–induced corneal neovascularization and reduce need for transplantation: the I–CAN study. Ophthalmology. 2014;121(9):1683‐92. 29.Lorenz K, Scheller Y, Bell K, Grus F, Ponto KA, Bock F, et al. A prospective, randomised, placebo–controlled, double–masked, three–armed, multicentre phase II/III trial for the Study of a Topical Treatment of Ischaemic Central Retinal Vein Occlusion to Prevent Neovascular Glaucoma – the STRONG study: study protocol for a randomised controlled trial. Trials. 2017;18(1):128. 30.Pfeiffer N, Voykov B, Renieri G, Bell K, Richter P, Weigel M, et al. First–in–human phase I study of ISTH0036, an antisense oligonucleotide selectively targeting transforming growth factor beta 2 (TGF–β2), in subjects with open–angle glaucoma undergoing glaucoma filtration surgery. PLoS One. 2017;12(11):e0188899. 31.Naert T, Colpaert R, Van Nieuwenhuysen T, Dimitrakopoulou D, Leoen J, Haustraete J, et al. CRISPR/Cas9 mediated knockout of rb1 and rbl1 leads to rapid and penetrant retinoblastoma development in Xenopus tropicalis. Sci Rep. 2016;6:35264. 32.Wu WH, Tsai YT, Justus S, Lee TT, Zhang L, Lin CS, et al. CRISPR Repair Reveals Causative Mutation in a Preclinical Model of Retinitis Pigmentosa. Mol Ther. 2016;24(8):1388–94. 33.Bakondi B, Lv W, Lu B, Jones MK, Tsai Y, Kim KJ, et al. In Vivo CRISPR/Cas9 Gene Editing Corrects Retinal Dystrophy in the S334ter–3 Rat Model of Autosomal Dominant Retinitis Pigmentosa. Mol Ther. 2016;24(3):556‐63. 34.Burnight ER, Giacalone JC, Cooke JA, Thompson JR, Bohrer LR, Chirco KR, et al. CRISPR–Cas9 genome engineering: Treating inherited retinal degeneration. Prog Retin Eye Res. 2018;65:28‐49. 35.Huang KC, Wang ML, Chen SJ, Kuo JC, Wang WJ, Nhi Nguyen PN, et al. Morphological and Molecular Defects in Human Three–Dimensional Retinal Organoid Model of X–Linked Juvenile Retinoschisis. Stem Cell Reports. 2019;13(5):906–23. 36.Kim EK, Kim S, Maeng YS. Generation of TGFBI knockout ABCG2+/ABCB5+ double–positive limbal epithelial stem cells by CRISPR/Cas9–mediated genome editing. PLoS One. 2019;14(2):e0211864. 37.Kim K, Park SW, Kim JH, Lee SH, Kim D, Koo T, et al. Genome surgery using Cas9 ribonucleoproteins for the treatment of age–related macular degeneration. Genome Res. 2017;27(3):419‐426.Crossref PubMed 38.Peddle CF, MacLaren RE The Application of CRISPR/Cas9 for the Treatment of Retinal Diseases. Yale J Biol Med. 2017;90(4):533‐41. 39.Ruan GX, Barry E, Yu D, Lukason M, Cheng SH, Scaria A. CRISPR/Cas9–Mediated Genome Editing as a Therapeutic Approach for Leber Congenital Amaurosis 10. Mol Ther. 2017;25(2):331‐41. 40.Suzuki K, Tsunekawa Y, Hernandez–Benitez R, Wu J, Zhu J, Kim EJ, et al. In vivo genome editing via CRISPR/Cas9 mediated homology–independent targeted integration. Nature. 2016;540(7631):144‐9. 41.Taketani Y, Kitamoto K, Sakisaka T, Kimakura M, Toyono T, Yamagami S, et al. Repair of the TGFBI gene in human corneal keratocytes derived from a granular corneal dystrophy patient via CRISPR/Cas9–induced homology–directed repair. Sci Rep. 2017;7(1):16713. 42.Xu CL, Park KS, Tsang SH. CRISPR/Cas9 genome surgery for retinal diseases. Drug Discov Today Technol. 2018;28:23‐32. 43.Yu W, Mookherjee S, Chaitankar V, Suja Hiriyanna, Kim J–W, Brooks M, Ataeijannati Y, et al. Nrl knockdown by AAV–delivered CRISPR/Cas9 prevents retinal degeneration in mice. Nat Commun. 2017;8:14716. 44.Zhu J, Ming C, Fu X, Duan Y, Hoang DA, Rutgard J, Zhang R, et al. Gene and mutation independent therapy via CRISPR–Cas9 mediated cellular reprogramming in rod photoreceptors. Cell Res. 2017;27:830–3. 45.Chung SH, Mollhoff IN, Nguyen U, Nguyen A, Stucka N, Tieu E, et al. Factors Impacting Efficacy of AAV–Mediated CRISPR–Based Genome Editing for Treatment of Choroidal Neovascularization. Mol Ther Methods Clin Dev. 2020;17:409‐17. 46.Huang X, Zhou G, Wu W, Duan Y, Ma G, Song J, et al. Genome editing abrogates angiogenesis in vivo. Nat Commun. 2017;8(1):112. 47.Yiu G, Tieu E, Nguyen AT, Wong B, Smit–McBride Z. Genomic Disruption of VEGF–A Expression in Human Retinal Pigment Epithelial Cells Using CRISPR–Cas9 Endonuclease. Invest Ophthalmol Vis Sci. 2016;57(13):5490–7. 48.Lightfoot JD, Fuller KK. CRISPR/Cas9–Mediated Gene Replacement in the Fungal Keratitis Pathogen Fusarium solani var. petroliphilum. Microorganisms. 2019;7(10):457. 49.Yang TC, Chang CY, Yarmishyn AA, Mao YS, Yang YP, Wang ML, et al. Carboxylated nanodiamond–mediated CRISPR–Cas9 delivery of human retinoschisis mutation into human iPSCs and mouse retina. Acta Biomater. 2020;101:484–94. 50.Li F, Hung SSC, Mohd Khalid MKN, Wang J–H, Chrysostomou V, Wong VHY, et al. Utility of Self–Destructing CRISPR/Cas Constructs for Targeted Gene Editing in the Retina. Hum Gene Ther. 2019;30(11):1349‐60. 51.Schaefer KA, Wu WH, Colgan DF, Tsang SH, Bassuk AG, Mahajan VB. Unexpected mutations after CRISPR–Cas9 editing in vivo. Nat Methods. 2017;14(6):547‐48. 52.Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search–and–replace genome editing without double–strand breaks or donor DNA. Nature. 2019;576(7785):149–57. 53.Hernandez M, Recalde S, Garcia–Garcia L, Bezunartea J, Miskey C, Johnen S, Diarra S, et al. Preclinical Evaluation of a Cell–Based Gene Therapy Using the Sleeping Beauty Transposon System in Choroidal Neovascularization. Mol Ther Methods Clin Dev. 2019;15:403–17. Financial disclosure: No author has a financial or proprietary interest in any material or method mentioned. No conflict of interest is declared.
|