J.ophthalmol.(Ukraine).2022;2:57-62.
|
http://doi.org/10.31288/oftalmolzh202225762 Received: 07 December 2021; Published on-line: 30 April 2022 Ehlers–Danlos syndrome: a case report M. M. Umanets, O. V. Zborovska, O. E. Dorokhova, M. L. Kogan SI "The Filatov Institute of Eye Diseases and Tissue Therapy of the NAMS of Ukraine"; Odesa (Ukraine) E-mail: mihailkogan2@gmail.com TO CITE THIS ARTICLE: Umanets MM, Zborovska OV, Dorokhova OE, Kogan ML. Ehlers–Danlos syndrome: a case report. J.ophthalmol.(Ukraine).2022;2:57-62. http://doi.org/10.31288/oftalmolzh202225762 Background: Ehlers–Danlos syndrome (EDS) is a rare disease characterized by connective tissue dysplasia, and, consequently, structural changes in ocular tissues. To the best of our knowledge, only 3 cases of surgical treatment with vitrectomy for retinal detachment in EDS have been reported in the literature. Purpose: To review an approach to medical and surgical treatment for recurrent retinal detachment in a patient with EDS. Material and Methods: Data on the clinical condition of eyes, examination findings and outcomes of conservative and surgical treatment were reported. Results: Substantial remnants of the vitreous were found in the periphery during surgical re-examination of the vitreous cavity, and were as much as possible removed with a vitreous cutter. Trypan blue staining of the retina facilitated the removal of the epiretinal membrane covering the whole posterior pole, and the membrane was completely removed with forceps. Conclusion: We argue for a more differentiated approach to the examination and treatment of patients with Ehlers–Danlos syndrome, because connective tissue dysplasia may mimic another systemic autoimmune disease. Early treatment and adequate treatment strategy enable treatment success for especially severe cases. Keywords: Ehlers–Danlos syndrome, leptoscleria, retinal detachment, vitrectomy
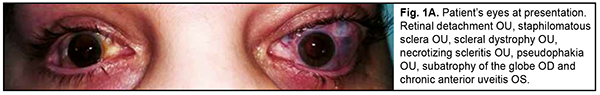
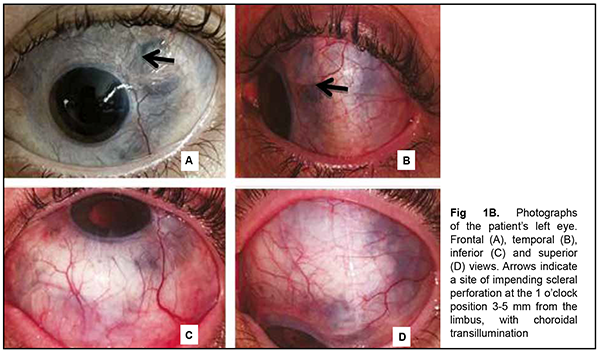
Ehlers–Danlos syndrome (EDS) is a rare disease characterized by connective tissue dysplasia, joint hypermobility, skin hyperextensibility, vascular fragility, and variable clinical picture [1]. The above manifestations are caused by defective collagen structure or abnormal collagen synthesis. The syndrome takes its name from dermarologists Edvard Ehlers and Henri-Alexandre Danlos. The hallmarks of the disorder include: (1) characteristic craniofacial features, (2) congenital multiple contractures, including adducted thumbs and talipes equinovarus, (3) characteristic cutaneous features including fine palmar creases, (4) peculiar finger shapes, (5) progressive spinal and foot deformities, (6) large subcutaneous hematomas, and (7) ophthalmological and urogenital involvement [2]. The famous violin virtuoso Nicolo Paganini had hypermobile joints, a slender physique, thoracic deformity and joint laxity, all features consistent with EDS [3]. Material and Methods We report a case of ophtalmological involvement in a 24 year-old female patient with EDS (Fig. 1). She presented to the Filatov institute and was initially diagnosed with scleral dystrophy OU, necrotizing scleritis OU, retinal detachment OU, and pseudophakia OU; subatrophy of the globe OD; and chronic anterior uveitis OS. The patient reported that, in 2018, she received transciliary vitrectomy with air-gas tamponade for retinal detachment in both eyes in the USA. However, her medical records regarding the diagnosis and received therapy were unavailable. Because she reported that after transciliary vitrectomy she was administered prednisolone orally for a year, there was a question whether the detachment was exudative, and whether it was accompanied by uveitis at the time of patient’s first visit to an ophthalmologist.
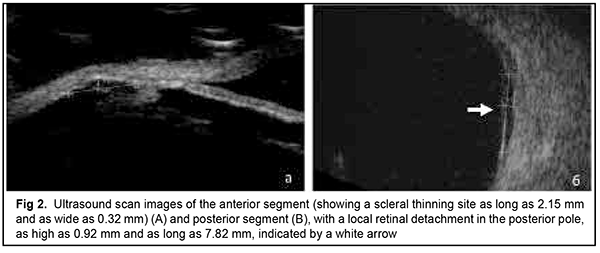
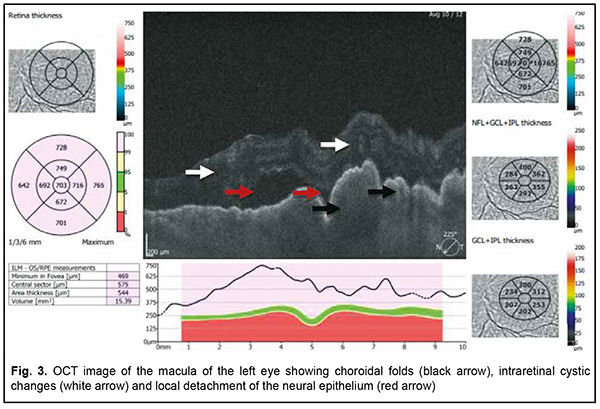
The patient also reported that her visual acuity was high immediately after surgery until June, 2020, with a subsequent gradual reduction in visual acuity in both eyes. She was diagnosed with complicated cataract and proposed to have lens phacoemulsification and intraocular lens (IOL) implantation after being examined by a local ophthalmologist. The operation in the left eye was successfull, but expulsive hemorrhage developed intraoperatively in the left eye, resulting in loss of light perception and subsequent phthisis bulbi. After she noted a significantly decreased vision in her left eye in May, 2021, she was referred to the Filatov institute, a tertiary eye care institution, for treatment. There she was urgently hospitalized and initially diagnosed with scleral dystrophy OU, necrotizing scleritis OU, retinal detachment OU, and pseudophakia OU; subatrophy of the globe OD; and chronic anterior uveitis OS. At presentation, there was loss of light perception OD and visual acuity was 0.05 with a spherical correction of -2.5 D. Intraocular pressure (IOP) as assessed by Maklakoff tonometer was 8-9 mmHg OU. The retinal detachment was localized in the posterior segment, but indirect ophthalmoscopy by an expert failed to visualize retinal tears. After laser photocoagulation, chorioretinal adhesions were noted at the periphery from 8 to 4 o’clock, but laser photocoagulation was not performed in the inferior segment of the eye. Given that the anterior uveitis was confirmed by the Kowa FM-600 laser flare meter (Kowa Company Ltd., Nagoya, Japan) with a flare level of 147.6(7.6) ph/ms and because of the history data, it could not be excluded that the nature of the retinal detachment was exudative. In addition, it is noteworthy that although the ocular lesions were severe, the changes in other organs and systems were not significant. The only echocardiographic evidence of abnormality was evidence of mitral valve prolapse. The patient also noted that a hemorrhagic rash appeared periodically on her lower legs and was followed by pigmentation. Given the above-mentioned, the scleral lesion was considered not only as a manifestation of Ehlers–Danlos syndrome, but also as potential necrotizing scleritis. Necrotizing scleritis frequently accompanies life-threatening autoimmune disorders like rheumatoid arthritis and systemic vasculitides (e.g., granulomatosis with polyangiitis and systemic lupus erythematosus (SLE)) [10-13]. Additional tests (ANA screen, ANA profile, ANCA screen, anti-cyclic citrullinated peptide (anti-CCP) assay, antiphospholipid syndrome profile, and lupus anticoagulant assay) were administered to exclude or confirm systemic autoimmune disorders. The patient was positive for both anti-Rib-P autoantibodies and lupus anticoagulant. The presence of antibodies against ribosomal P proteins has been found to be very specific for patients with SLE compared with either healthy controls or with controls who had other rheumatic diseases [12]. In addition, Carmona-Fernandes and colleagues [13] found that, of the patients with SLE who had tested positive for anti-Rib-P, most were positive for anti-Rib-P plus antibodies against double-stranded DNA (anti-dsDNA) [13]. The patient was referred to the rheumatologist. However, due to the lack of clinical manifestations, the diagnosis of SLE was not confirmed but could not be definitely excluded at the time of examination by rheumatologist. Given the severity of the ocular lesions, we decided, in conjunction with the rheumatologist, to administer immunosuppressive therapy (methylprednisolone 24 mg daily, with a subsequent reduction in daily dosage, and methotrexate 7.5 mg weekly). Imaging included B-scan and ocular coherence tomography (OCT). B-scan ultrasonography of the anterior and posterior segments found scleral thinning (with a sclera as thin as 0.1 mm) along an entire circumference of the sclera (Fig. 2). There was evidence of a 0.3 mm prolapse of the globe outwards along 2.2 mm in the paralimbal region. In addition, B-scan of the left eye demonstrated a moderately echogenic membranous structure (as high as 0.9 mm and as long as 5.9 х 7.8 mm) that was marginally adhered to the vitreoretinal interface at 10 o’clock at the extreme periphery of the retina, with retinal adherence in other directions. OCT showed (a) choroidal folds due to hypotony and (b) intraretinal cystic changes with local detachment of the neural epithelium (Fig. 3).
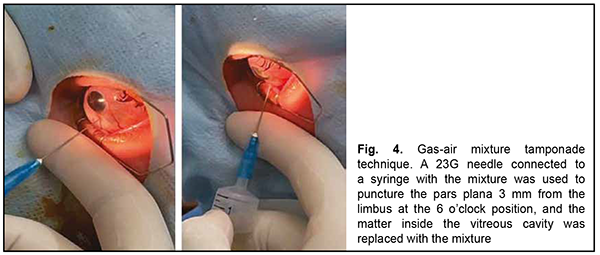
After a course of conservative treatment (including a course of immunosuppressive therapy), best-corrected visual acuity OS improved to 0.5 and the exudative detachment decreased in extent. The IOP increased and ocular hypertension developed as early as day 4 of treatment, which was treated by antihypertensive eye drops. Uveitis activity decreased, but the sclera almost did not change in the presence of administered anti-inflammatory therapy. Apparent choroidal prolapse at areas of choroidal thinning was noted. Due to the reasons identified above, the management strategy for our patient was revised. Because of an anti-scarring effect of immunosuppressive agents, and in the absence of a confirmed final systemic autoimmune diagnosis, it was decided to withdraw methotrexate, and to reduce the daily dose of methylprednisolone 2 mg/day. In addition, in an attempt to improve the state of the connective tissue, the patient was prescribed dietary supplements containing collagen and silicone for 45 days. However, 4 days after discharge, the visual acuity in the left eye again decreased to several hundredths. A high retinal detachment with macular involvement was noted in the fundus, but an expert failed to visualize retinal tears. Given the risk of the development of blindness risk due to iatrogenic scleral damage, it was decided to perform a less traumatic surgical procedure, namely, replacement tamponade with a mixture of perfluoropropane (C3F8) and air for retinopexy. In brief, a 23G needle connected to a syringe with the mixture was used to puncture the pars plana 3.5 mm from the limbus at the 6 o’clock position with a maximally dilated pupil (Fig. 4). Vitreous fluid was aspirated and replaced by the air-perfluoropropane gas mixture until a normal intraocular pressure was obtained. It is noteworthy that as early as the next day after replacement tamponade, a complete retinal attachment was achieved, and a gas bubble filled 95-100% of the vitreous cavity. At day 3 after replacement tamponade, a complete retinal attachment was achieved, and, to form chorioretinal adhesion in the inferior fundus, transpupillary retinal laser photocoagulation was performed using a 532-nm PUREPOINT laser (Purepoint Laser, Alcon Laboratories, Inc., Fort Worth, TX, USA) connected to a binocular indirect ophthalmoscope (Omega 500, Heine Optotechnik, Herrshing, Germany). The laser connected to a binocular indirect ophthalmoscope enables performing retinal laser photocoagulation even in the presence of gas-air endotamponade.
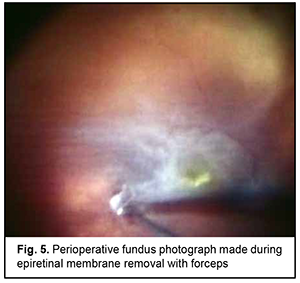
At 2 weeks, as the air-perfluoropropane gas mixture resolved, a recurrent retinal detachment in the upper outer quadrant and the inferior half of the fundus due to the development of proliferative retinopathy was noted. Although the patient did maintain the prescribed head position in an attempt to make the gas bubble remaining in the same, correct position within the eye, this resulted in no effect on the detached retina. In this connection, the patient was proposed to have a three-port surgical re-examination of the vitreous cavity via a transciliary approach. This was a 25-G surgery with the use of a Constellation system (Alcon) and a wide-field OMS-800 OFFISS (Topcon, Japan) operating microscope. In this procedure, the surgeon should be very careful to avoid iatrogenic scleral damage while using a vitreous instrument to turn the globe or while increasing infusion fluid pressure. Substantial remnants of the vitreous were found in the periphery during surgical re-examination of the vitreous cavity, and were as much as possible removed with a vitreous cutter. Trypan blue staining facilitated the removal of the epiretinal membrane (ERM) covering the whole posterior pole, with the membrane completely removed with forceps (Fig. 5). Because the patient had apparent manifestations of proliferative vitreoretinopathy (fixed retinal folds and intraretinal fibrosis), a 180-degree retinotomy in the inferior retina was created, perfluorodecalin was used to unfold and flatten the retina, laser photocoagulation burns were applied along the margins of the retinotomy, and silicone oil-RMN3 (Oxane HD) silicone oil (Oxane HD) vitreous tamponade was performed. Although small-gauge (25G) instrumentation was utilized, sclerotomies required 8-0 nylon suture placement due to marked scleral thinning.
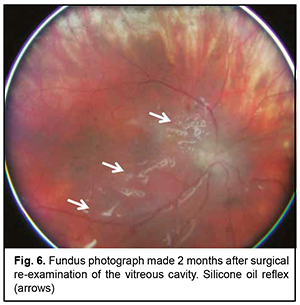
Postoperatively, special attention was given to daily monitoring of IOP, visual functions, and state of staphilomatous scleral sites. We achieved anatomic retinal attachment, with an improvement in visual acuity to 0.05 OS. It is noteworthy that although the anterior hyaloid membrane and posterior IOL capsule were removed during surgical re-examination of the vitreous cavity, there was apparent pigment accumulation on the posterior IOL surface in the early postoperative period. At 3 months after surgery, there was ophthalmoscopic evidence of reduced pigment accumulation on the posterior IOL surface (Fig. 6), and visual acuity improved to 0.3 OS. In addition, the patient showed normal IOP without ocular hypotensive treatment. Moreover, uveitis activity decreased significantly (a flare level decreased to 39.1 (1.9) ph/ms).
Discussion Characteristic ocular manifestations of EDS include leptoscleria (i.e., scleral thinning), changes in the cornea (microcornea, keratoglobus and keratoconus,), corneal hydrops, spontaneous ruprure of the cornea and/or sclera, xerophthalmia, high myopia, inadequate convergence, lens subluxation, retinal detachment, glaucoma, and recurrent vitreous hemorrhage [6, 7]. The International EDS Consortium has proposed a revised EDS classification, which recognizes 13 subtypes. However, in view of the vast genetic heterogeneity and phenotypic variability of the EDS subtypes, and the clinical overlap between EDS subtypes, but also with other heritable connective tissue disorders, the definite diagnosis of all EDS subtypes, except for the hypermobile type, relies on molecular confirmation with identification of causative genetic variant(s) [5]. To the best of our knowledge, there have been only 3 reports (those by Bodanowitz and colleagues (1997) [7], Whitlow and colleagues (2019) [8], and Lumi and colleagues [9]) of surgical treatment of retinal detachment in EDS by vitrectomy. Bodanowitz and colleagues [7] described several complications during the surgery, such as choroidal detachment and intensive bleeding. The closure of the sclerotomies was also difficult due to the very thin sclera. Because of redetachment with proliferative vitreoretinopathy, the patient needed two revitrectomies. The final visual acuity after the last surgical procedure with silicon oil tamponade was 6/60. Whitlow and colleagues [8] also noted that recurrent retinal detachments were caused by PVR, and the patient required several vitrectomy reoperations with silicone oil tamponade of the vitreous cavity. Treatment of patients with leptoscleria (i.e., scleral thinning) is associated with numerous potential perioperative complications (e.g., damage to the scleral capsule). Whitlow and colleagues [8] described vitrectomy with additional donor scleral graft suturing to the patient's sclera. They achieved anatomic retinal attachment, with an improvement in visual acuity to 1/60 in the affected eye [8]. Unfortunately, in our case, it did not seem possible to use a scleral autograft because of a large scleral area as thin as 0.1 mm. Lumi and colleagues [9] reported on good anatomical and functional outcome of 25G vitrectomy completed with gas endotamponade for retinal detachment in a patient with EDS type, with a follow-up of 8 months. Mild postoperative vitreous hemorrhage was resorbed in first week after the surgery. Postoperative extensive pigment dispersion on the posterior lens face persisted for several weeks [9]. In our patient, there was apparent pigment accumulation on the posterior IOL surface in the early postoperative period, but, at 3 months after surgery, there was ophthalmoscopic evidence of reduced pigment accumulation on the posterior IOL surface, and visual acuity improved to 0.3 OS. In addition, we managed to (1) avoid iatrogenic scleral damage and scleral grafting and (2) achieve good anatomical and functional outcome in a patient with a single eye. Conclusion We argue for a more differentiated approach to the examination and treatment of patients with Ehlers–Danlos syndrome, because connective tissue dysplasia may mimic another systemic autoimmune disease. Early treatment and adequate treatment strategy enable treatment success for especially severe cases. We managed to achieve anatomic retinal attachment and to improve vision in our patient with a single eye in spite of impending scleral perforation. Conflict of Interest Statement. The authors declare no conflict of interest. Funding Support. There are no external sources of funding. References Athor Contribution. A.V. Zborovskaya: conservative treatment of a patient, concept, and editing; A.E. Dorokhova: conservative treatment of a patient; N.N. Umanets: surgical treatment, the idea for the publication and the editing of the article; M.B. Kogan: literature analysis and article formation.
References Conflict of Interest Statement. The authors declare no conflict of interest. Funding Support. There are no external sources of funding. References Athor Contribution. A.V. Zborovskaya: conservative treatment of a patient, concept, and editing; A.E. Dorokhova: conservative treatment of a patient; N.N. Umanets: surgical treatment, the idea for the publication and the editing of the article; M.B. Kogan: literature analysis and article formation.
References 1.Vit VV. [Pathology of the eye, ocular adnexa and orbit]. Vol. 1. Odesa:Astroprint; 2019. Russian. 2.Brady AF, Demirdas S, Fournel-Gigleux S, et al. The Ehlers-Danlos syndromes, rare types. Am J Med Genet C Semin Med Genet. 2017 Mar;175(1):70-115. 3.McKusick VA. Heritable disorders of connective tissue. 4th ed. St. Louis, MO: CV Mosby; 1972. 4.Villani E, Garoli E, Bassotti A, Magnani F, Tresoldi L, Nucci P, et al. The cornea in classic type Ehlers–Danlos syndrome: macro- and microstructural changes. Invest Ophthalmol Vis Sci. 2013 Dec 11;54(13):8062-8. 5.Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, et al. The 2017 international classification of the Ehlers–Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017 Mar;175(1):8-26. 6.Perez-Roustit S, Nguyen DT, Xerri O, Robert M, De Vergnes N, Mincheva Z, et al. Ocular manifestations in Ehlers–Danlos Syndromes: clinical study of 21 patients. J Fr Ophtalmol. 2019 Sep;42(7):722-9. 7.Bodanowitz S, Hesse L, Pöstgens H, Kroll P. Retinal detachment in Ehlers–Danlos syndrome. Treatment by pars plana vitrectomy. Ophthalmologe. 1997 Sep;94(9):634-7. 8.Whitlow S, Idrees Z. RD repair using 360-degree scleral graft for extensive scleral ectasia in a patient with Ehlers–Danlos syndrome. Am J Ophthalmol Case Rep. 2019 Sep 13;17:100554. 9.Lumi X, Bergant G, Lumi A, et al. Outcomes of vitrectomy for retinal detachment in a patient with Ehlers–Danlos syndrome type IV: a case report. J Med Case Reports. 2021 May 20;15(1):249. 10.Hernández-Camarena JC, Rodríguez-García A., Valdez-García J. Diagnosis and treatment approach for necrotizing scleritis (NS): a clinical case. Gac Med Mex. Jul-Aug 2015;151(4):525-8. 11.Ashok D, Ayliffe WH, Kiely PDW. Necrotizing scleritis associated with rheumatoid arthritis: long-term remission with high-dose infliximab therapy. Rheumatology (Oxford). 2005 Jul;44(7):950-1. 12.Sainz de la Maza M, Foster CS, Jabbur NS. Scleritis associated with systemic vasculitic diseases. Ophthalmology. 1995 Apr;102(4):687-92.. 13.Carmona-Fernandes D, Santos MJ, Canhão H, et al. Anti-ribosomal P protein IgG autoantibodies in patients with systemic lupus erythematosus: diagnostic performance and clinical profile. BMC Med. 2013 Apr 4;11:98.
|